How to Sequence RRBS Libraries on Illumina® Platforms
What is RRBS?
There is an ever-growing number of NGS methods available to profile DNA methylation. Reduced representation bisulfite sequencing (RRBS) is an established and cost-efficient method that is well-suited for genome-wide DNA methylation profiling. RRBS utilizes restriction digest to enrich for CpG dense regions. The restriction enzyme MspI is methylation insensitive which cuts at cytosines in a CpG context (Figure 1). Therefore, by digesting samples with MspI, it is guaranteed that each fragment will contain at least two CpG sites. Because DNA methylation occurs primarily in a CpG context, sequencing only CpG rich regions allows researchers to capture large amounts of methylation data at a reduced cost. Unlike other enrichment methods, RRBS can profile samples at a single base resolution, making it an excellent technique for pilot methylation studies.
RRBS Library Characteristics
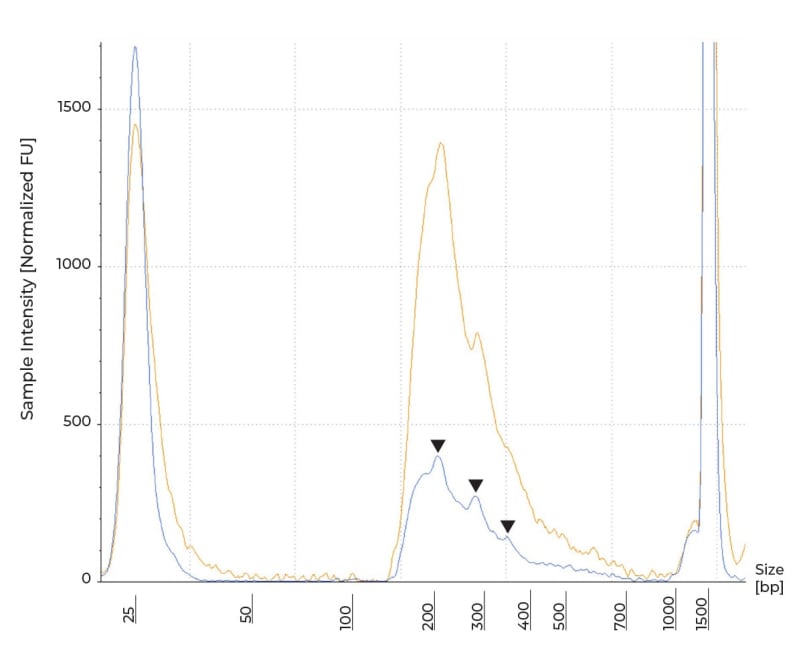
Most library preparation protocols require fragmentation of the input to achieve the appropriate insert size. As such, a successful library will have a fragment profile with a peak(s) corresponding to the expected insert. In RRBS, MspI simultaneously enriches for CpG dense regions and fragments the DNA, resulting in shorter libraries compared to other common libraries such as RNA-seq. Because MspI recognizes a specific cut site (Figure 1), the resulting library will have a set pattern determined by the distribution of the cut site across the genome from individual species. In human samples, MspI digestion will results in peaks at around 69, 134 and 205 bp, as described by Oda et al.[1] Following MspI digestion, the spikes correspond to the fragments’ length plus full-length Illumina TruSeq adapters’ length.
How to Sequence RRBS Libraries
Library Concentration and Quantification
As with all library preparations, quantification with a qPCR-based method such as KAPA Quantification for Illumina, or similar, is critical to ensuring an accurate loading concentration. Because RRBS libraries are comprised of a range of fragment sizes, some of which can be relatively small, the size-adjustment calculation is necessary following KAPA quantification. Larger fragments will produce a greater fluorescent signal compared to shorter fragments with the same concentration – thus, if the fragments are longer or shorter than that of the DNA standard, size-adjustment must be performed. In addition, it is prudent to perform KAPA quantification on the PhiX standard alone after receiving it to confirm the appropriate amount of spike-in is used.

PhiX Spike-In
RRBS libraries require special considerations for sequencing: as a result of CpG enrichment and bisulfite conversion, RRBS libraries are AT rich and therefore low complexity, in addition to a set pattern at the beginning of each library insert due to MspI digestion. The commonly used PhiX spike-in control can not only monitor critical NGS metrics, but also introduce library diversity, such as in RRBS. The optimal amount of PhiX spike-in for RRBS libraries will vary depending on the contents of the sequencing pool as well as the specific Illumina sequencing platform. Below are suggested starting points for PhiX using the NovaSeq. Further optimization on the PhiX spike-in percentage may be necessary based on the sequencing metrics of the first run.
| Library Pool Composition | Suggested % PhiX |
|---|---|
| Low Complexity Libraries <50% | PhiX optional, or follow Illumina’s general guidance |
| 50% |
5% |
| 100% Low Complexity Libraries | >5% |
Preparing the Final Library
Once the libraries and spike-in are quantified, they should be diluted and denatured according to the system-specific guides provided by Illumina. A simple method for preparing the spike-in is to first dilute the library pool and the PhiX to the same concentration, then combine corresponding volumes to reach the expected spike-in percentage. For example, to achieve a 10% PhiX spike-in at a loading concentration of 1.6 pM with a total volume of 1300 µL of the combined library and PhiX control, dilute the library pool and the PhiX to 1.6 pM, respectively, and then combine 1170 µL of the library pool with 130 µL of the PhiX. Due to the small size of RRBS libraries compared to other library types, a lower loading concentration than the recommended value in Illumina’s standard guide may be necessary to avoid over-clustering.
Conclusions
RRBS is a cost-effective, genome-wide DNA methylation profiling technique. For best results, special attention should be paid to library quantification and spike-in parameters.
LEARN MORE ABOUT ZYMO RESEARCH'S RRBS WORKFLOW
Learn MoreReferences
- Mayumi Oda, Jacob L. Glass, Reid F. Thompson, Yongkai Mo, Emmanuel N. Olivier, Maria E. Figueroa, Rebecca R. Selzer, Todd A. Richmond, Xinmin Zhang, Luke Dannenberg, Roland D. Green, Ari Melnick, Eli Hatchwell, Eric E. Bouhassira, Amit Verma, Masako Suzuki, John M. Greally, High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers, Nucleic Acids Research, Volume 37, Issue 12, 1 July 2009, Pages 3829–3839, https://doi. org/10.1093/nar/gkp260.





