Successfully Added to Cart
Customers also bought...

-
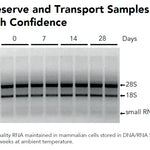 DNA/RNA Shield (50 ml)Cat#: R1100-50DNA/RNA Shield reagent is a DNA and RNA stabilization solution for nucleic acids in any biological sample. This DNA and RNA stabilization solution preserves the...
DNA/RNA Shield (50 ml)Cat#: R1100-50DNA/RNA Shield reagent is a DNA and RNA stabilization solution for nucleic acids in any biological sample. This DNA and RNA stabilization solution preserves the... -
 DNA/RNA Shield SafeCollect Swab Collection Kit (1 ml fill) (1 collection kit)Cat#: R1160The DNA/RNA Shield SafeCollect Swab Collection Kit is a user-friendly collection kit for stabilizing the nucleic acid content of samples collected with a swab. DNA/RNA...
DNA/RNA Shield SafeCollect Swab Collection Kit (1 ml fill) (1 collection kit)Cat#: R1160The DNA/RNA Shield SafeCollect Swab Collection Kit is a user-friendly collection kit for stabilizing the nucleic acid content of samples collected with a swab. DNA/RNA...


Highlights
- Fast: 80 - 90% of E.coli are lysed in only 10 minutes after harvesting.
- Simple 20 Second Transformation: No heat shock! Just add DNA and spread.
- DE3 Lysogen: Encodes the T7 polymerase for expressing recombinant proteins under the control of the T7 promoter.
Original Manufacturer
100% satisfaction guaranteed, read Our Promise
Innovated in California, Made in the USA
Highlights
- Fast: 80 - 90% of E.coli are lysed in only 10 minutes after harvesting.
- Simple 20 Second Transformation: No heat shock! Just add DNA and spread.
- DE3 Lysogen: Encodes the T7 polymerase for expressing recombinant proteins under the control of the T7 promoter.

Original Manufacturer
100% satisfaction guaranteed, read Our Promise
Innovated in California, Made in the USA
Description
Performance
Technical Specifications
| Autolysis | Lyses easily. The parent strain JM109 itself will release about 20% of cellular protein after one freeze-thaw cycle. This strain will lyse in a wide range of buffer conditions. 80-90% of E. coli are lysed after a single freeze-thaw treatment. |
|---|---|
| Cell Growth | Grows well, especially when medium is supplemented with 1 mM Mg2+ |
| DNA Extraction | This strain is EndA- and yields high quality DNA preparations. |
| DNA Stability | The RecA- mutation in XJa stabilizes repetitive DNA sequences. |
| Genotype | F`[traD36 proA+B+ laclq ∆(lacZ)M15] ∆(lac-proAB) glnV44 (supE44)e14- (McrA-) thi gyrA96 (NalR) endA1 hsdR17(rK- mK+) relA1 recA1 ΔaraB::λR, cat (CmR), λ(DE3) |
| Processing Time | 10 minutes |
| Product Storage | -70°C to -80°C |
| Protein Expression | Suitable for general screening, but proteases may degrade small or otherwise unstable recombinant proteins. |
| Transformation Efficiency | 108 - 109 transformants per µg of plasmid DNA |
Resources
Documents
FAQ
Do not perform the freeze and thaw cycle in a buffer containing glycerol. Glycerol protects the E.coli from forming ice crystals which are essential to the lysis of the cells.
Depending on the amount of material used, the lysed material may become viscous, preventing efficient manipulation. However, for most applications it is not necessary to use a large amount of cell material. If necessary, vortexing vigorously for 30 seconds will decrease viscosity in most cases. Alternatively, a nuclease treatment (e.g. DNAse I) can be used to reduce viscosity. Diluting the cell lysate with additional buffer will also reduce viscosity issues.
If the results obtained are not satisfactory, lysis can be significantly improved by incubating the cells at higher temperatures (25 - 37°C) or for longer time (10 or 20 minutes) after thawing (step 5).
Non-λ lysozyme usually is able to degrade chitin. However, the λ lysozyme expressed in these cells is not able to degrade chitin. λ lysozyme is a transglycosylase.
For best results, cells should not be growing actively prior to arabinose induction. This is achieved by using an overnight starter, where cells are already in the stationary growth phase, as stated in the protocol. If a fresher starter needs to be used, include arabinose already in the starter culture.
When glucose is added to the growth media, it inhibits the induction of the autolysis genes when it is present in the media. As the cells grow, they consume the glucose as a carbon source. Once the glucose has been consumed autolysis begins.
Resuspend the cell pellet in water with or without 0.01% - 0.1% Triton X-100. For His-tag purification, resuspend in the His-Binding Buffer of the His-spin Protein Miniprep kit (Zymo Research product # P2001 or P2002). Acidic buffers and buffers containing higher concentrations of Mg2+ (>1 mM), and related metals that stabilize cell walls, inhibit lysis reaction to a various extent. If possible, add magnesium to the buffer after cells are lysed.
All our competent cells are classified into Biosafety level 1 and are not genetic modified organisms. Only when transformed with a plasmid they become GMOs.
Most cloning strains will be dam+/dcm+ unless specifically noted in the genotype.
Yes
DH5α is equivalent to Zymo 5α. DH10B, Top10, and One Shot Top10 are equivalent to Zymo 10B. For XL-21 Blue, JM109 is the closest match and for Stbl3, HB101 is the closest match.
– Prepare fresh agar plates – Use more antibiotics in plates – Incubate plates for a shorter time after plating cells
We do not recommend diluting the competent cells. We recommend using less DNA to transform cells, or aliquot cells in smaller volumes before transformation. If absolutely necessary, cold 1X Competent Buffer (Mix & Go Transformation Kit, T3001 & T3002) should be used in the dilution.
No outgrowth is necessary when using Ampicillin or Carbenicillin for selection. However, an outgrowth step is required when using Chloramphenicol, Kanamycin, and Tetracycline because of the mode of action of the antibiotic itself. We recommend the following procedure for the outgrowth step: 1. Incubate cells on ice for 5-10 min after addition of plasmid. 2. Add 4 volumes of SOC media. 3. Incubate at 37°C for 60 min with gentle shaking at 200-300 rpm. 4. Spread on a pre-warmed culture plate containing the appropriate antibiotic.
For Zymo 5α and Zymo 10B up to 20kb. However, transformation efficiency decreases proportionally from 10-20kb. Above 20kb, cells are difficult to transform. JM109, HB101, XJa, XJa (DE3), XJb, XJb (DE3) and TG1 can handle constructs up to 10kb.
There really is no maximum or minimum recommended DNA concentration, but we use 10 pg for quality control. However, the volume of DNA added should not exceed 5% of the cells total volume; the efficiency can decrease several fold as the volume of DNA used increases. If the DNA sample is too diluted, use our DNA Clean & Concentrator.
1. Thaw cells on ice, not room temperature. 2. Incubate cells and DNA mixture on ice, not at room temperature. However, do not incubate longer then 1 hour. 3. Ensure cells are still frozen when received. 4. Pre-warm the culture plates at 37°C for at least 30 minutes. 5. Prepare fresh LB agar plates containing the appropriate antibiotic. 6. Prepare a new DNA sample. 7. Store the cells at -80°C (not 4°C or -20°C). If the freezer breaks, the cells should be OK as long as the temp does not go higher than -50°C. 8. Avoid freeze/thaw cycles.
Heat shock is not necessary, however sometimes it can be beneficiary when preparing libraries or transforming XJb Autolysis E. coli strains. We recommend the following protocol for Heat Shock with Outgrowth: 1. Incubate cells on ice for 5-10 min after addition of plasmid. 2. Incubate cells at 42°C for 45 seconds. 3. Add 450 ml of SOC to the cells. 4. Incubate at 37°C for 60 min with gentle shaking at 200-300 rpm. 5. Spread on a pre-warmed culture plate containing the appropriate antibiotic.
Citations
To clone new GFP-like fluorescent proteins from Obelia medusa, the authors identified the potential genes using expression libraries and cloned the genes into a vector. Expression of the proteins was facilitated by using XJb Autolysis E. coli cells from Zymo Research. The authors were able to purify three proteins from Obelia medusa that fluoresce in three different colors: cyan, green, and yellow.
Aglyamova, G.V. et al. (2011) Multi-colored homologs of the green fluorescent protein from hydromedusa Obelia sp. Photochem Photobiol Sci (8):1303-9.Need help? Contact Us