CONTROLS FOR DNA METHYLATION ASSAYS
How Controls Boost Reliability & Reproducibility
CONTROLS FOR METHYLATION ASSAYS: HOW, WHEN & WHY TO USE THEM
All scientific experiments need controls. Specific controls are required for certain type of experiments, such as DNA methylation assays. DNA methylation is a powerful, well-studied epigenetic mark that continues to provide extensive insight into transcription regulation, embryonic development, genome stability, and chromatin structure. Changes in DNA methylation profiles have also been linked to the development of many diseases 1. Many clinical applications have utilized DNA methylation as a biomarker for early detection, diagnostics, and personal therapy 2, 3. This requires accurate characterization of methylation patterns at a specific region or single-base level.
Most commonly used DNA methylation assays include bisulfite sequencing using NGS technologies, methylation-specific PCR (MSP), and methylation-sensitive restriction enzyme (MSRE) assays. To minimize bias in quantifying methylation levels, especially in clinical applications, each assay needs to be carefully designed and optimized to ensure its robustness4. Integrating established controls is key for optimizing assays and monitoring assay’s effectiveness . For optimization of DNA methylation assays, it is best to use established DNA standards with known methylation levels. Typically, this requires a methylated and non-methylated DNA standard, which contain nearly 100% and 0% methylation, respectively, at all CpG sites. The use of these two standards individually or in combination can help identify potential biases in assay development. For established workflows, these standards can serve as a positive and negative control to validate the procedures and results, which is essential in clinical testing to enhance quality control and assurance.
QUALITY CONTROLS FOR VALIDATING DNA METHYLATION ASSAY WORKFLOWS
As a good practice, methylated and non-methylated standards should be processed in parallel to experimental samples as a quality control for the whole workflow. In contrast to standards that are expected to produce a consistent result each time, experimental samples can have many variables, making troubleshooting difficult. In PCR-based and NGS-based DNA methylation assays, inhibitors carried over from the samples, primer design, enzymatic reagents, instrument failures, and many other factors can cause an assay to fail. Data produced by the standards can suggest where to start troubleshooting. For example, if standards perform as expected when run in parallel with experimental samples which perform poorly, the workflow is functioning properly, suggesting the experimental sample quality may be an issue. If standards did not perform as expected, there may be an issue within the workflow that caused the assay to fail.
CONTROLS FOR OPTIMIZING AND CALIBRATING BISULFITE-BASED ASSAYS
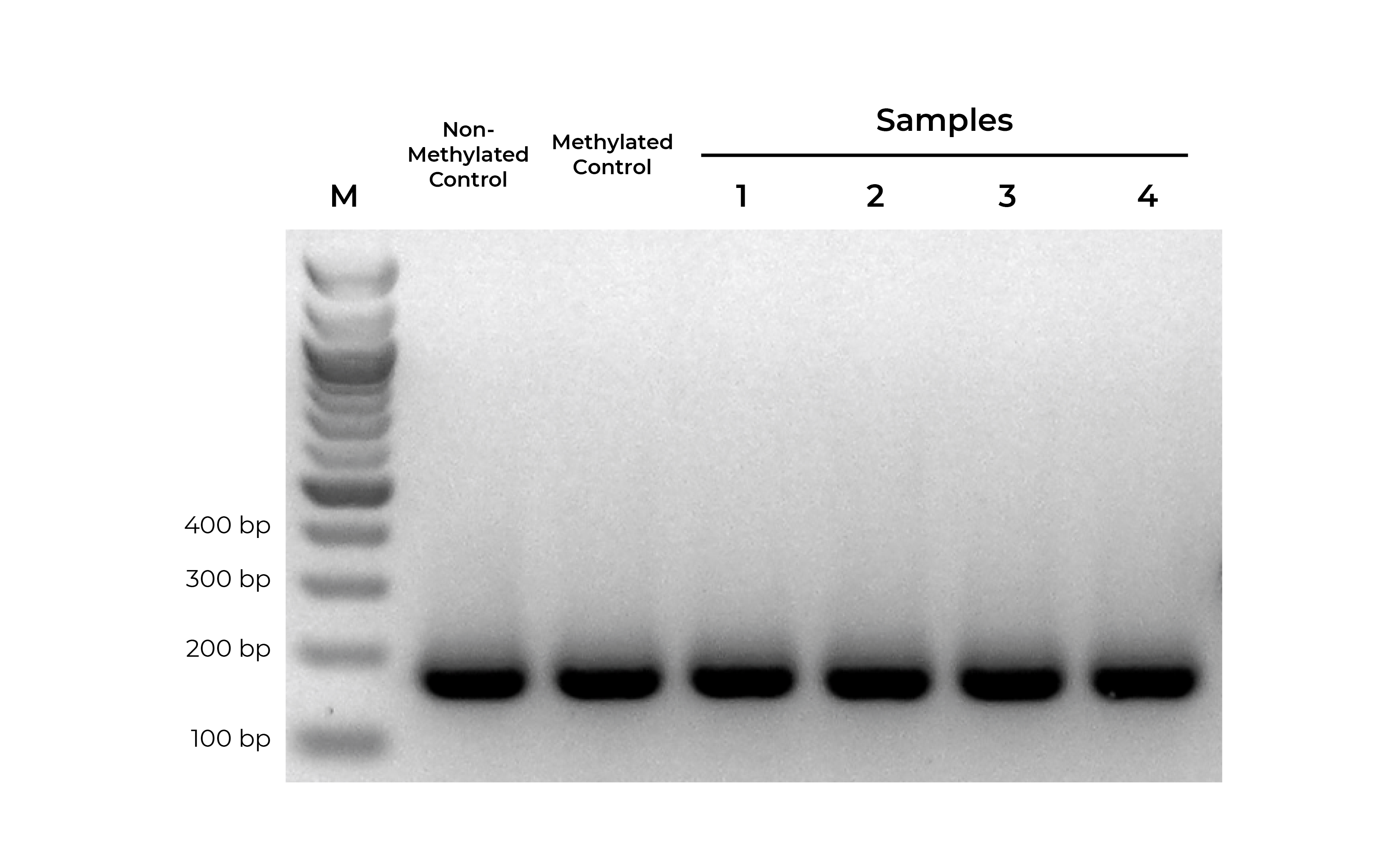
Bisulfite PCR (BSP) is a common methodology used for methylation analysis at single-base resolution. Samples are first bisulfite converted and amplified using bisulfite-specific primers. The PCR products generated are sequenced (i.e. Next-Gen Sequencing, Sanger Sequencing, pyrosequencing) and analyzed to determine the methylation status at every cytosine. However, it is critical that primer pairs designed for BSP amplify both methylated and non-methylated sequences with the same efficiency (Figure 1). Otherwise, any amplification bias can result in skewed methylation levels.
Using methylated and non-methylated DNA controls, BSP primers can be easily optimized (validated) based on following criteria:
- Specific product amplification
- Equally robust amplification of both methylated and non-methylated DNA standards
- When sequenced, the methylated and non-methylated DNA standards should result in nearly 100% and 0% methylation, respectively. If the results deviate from the expected result, this may indicate PCR bias.
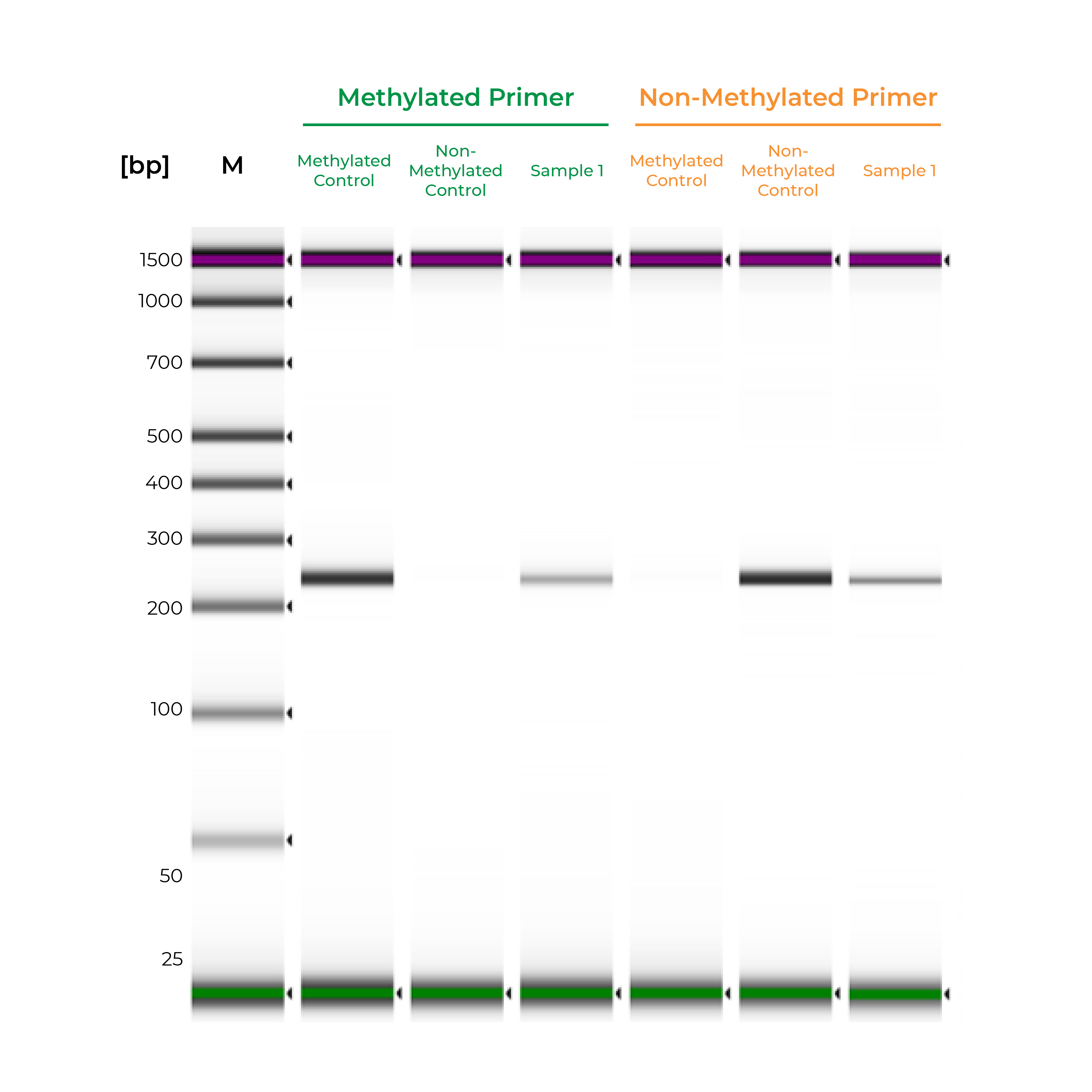
Unlike bisulfite PCR, methylation-specific PCR (MSP) requires methylated and non-methylated templates to be differentially amplified by two different primer sets. One is a methylation-specific primer set, and if amplification occurs, it indicates the amplified region is completely methylated. The second primer set targets non-methylated templates, so any amplification with this primer set indicates the amplified region is completely non-methylated. If amplicons are generated by both primer sets, the region is partially methylated.
Such two primer sets required for MSP can also be optimized and validated utilizing Methylated & Non-Methylated DNA Controls using following guidelines:
- Specific product amplification
- The methylated primer will only amplify the methylated control DNA and will not amplify the non-methylated DNA.
- The non-methylated primer will only amplify the non-methylated control DNA and will not amplify the methylated DNA.
During primer validation, having characterized methylated and non-methylated DNA standards can streamline the process. For example, if a slight PCR signal is observed using the methylated standard with the non-methylated primers, or vice versa, it would indicate that the primer set is non-specific. For established workflows, especially in clinical assays, methylated and non-methylated DNA standards can serve as positive and negative controls to validate the MSP workflow. Standards will help confirm that the entire process – bisulfite conversion, amplification, and analysis – are functioning appropriately, and the results for the test samples are reliable (Figure 2).
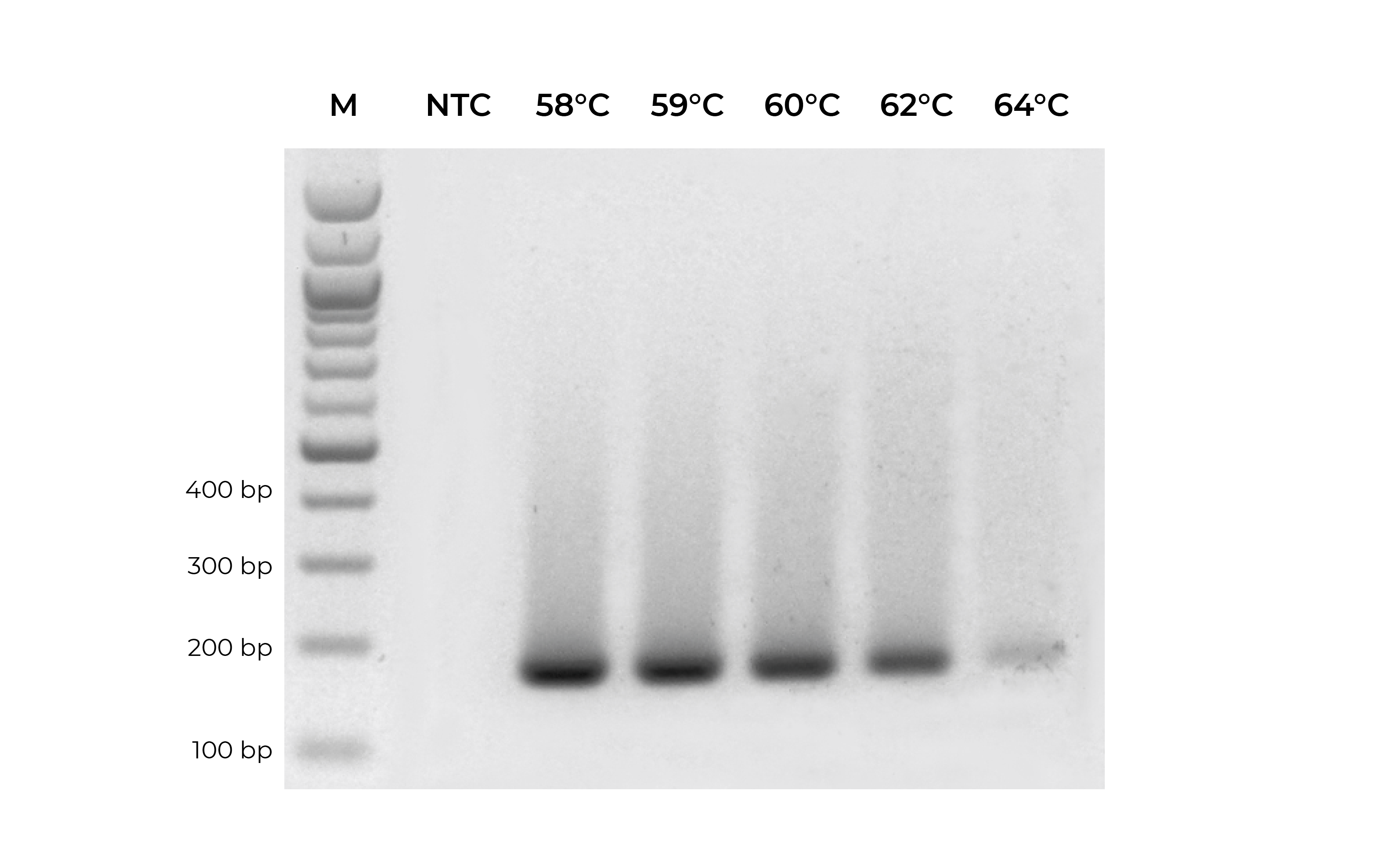
In addition to validating primers, DNA standards can be used in place of precious samples to optimize the annealing temperature for new primer sets (Figure 3). An annealing temperature gradient should be performed for each newly designed primer set to ensure optimal amplification of the intended target.
For more tips on primer design, see the Bisulfite Beginner Guide.
CONTROLS FOR METHYLATION SENSITIVE RESTRICTION ENZYME (MSRE) ASSAYS
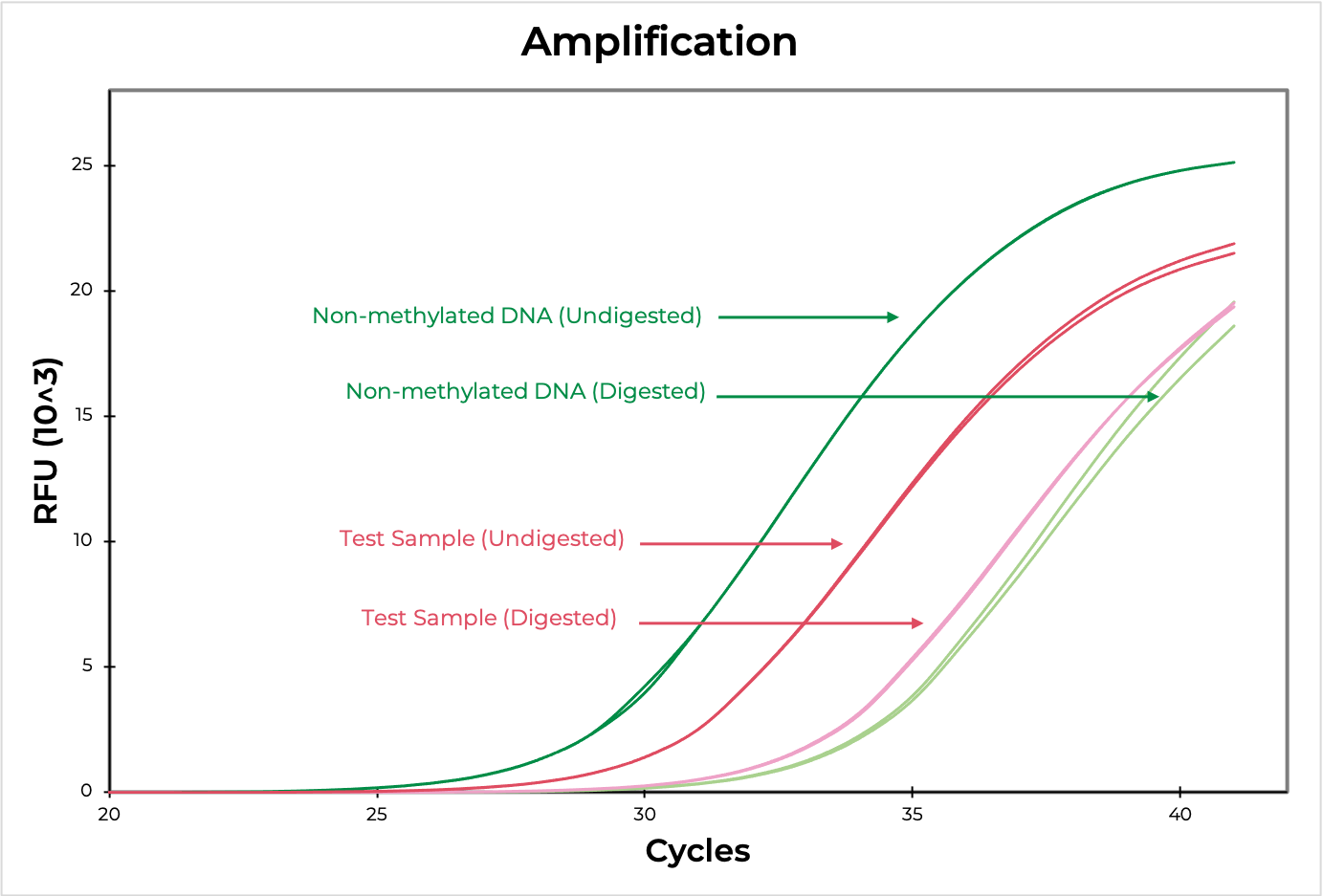
Methylation sensitive restriction enzyme (MSRE) assays rely on restriction enzymes being able to distinguish methylated versus non-methylated cytosines. If the restriction site is not methylated, the enzyme will digest the DNA while methylated sites will be uncut. Then, methylation at the site can be evaluated using quantitative PCR by comparing the amplification of the digested versus undigested sample. The smaller the ∆C(t) between the two, the higher the methylation levels are, and vice versa (Figure 4). DNA visualization methods, such as an agarose gel, can also be used to determine if the digestion occurred at the region of interest.
Methylated and non-methylated DNA standards should be digested in parallel to experimental samples to demonstrate the robustness of the restriction enzyme in distinguishing methylated versus non-methylated sites. This will also help determine the sensitivity of the assay to the region of interest.
DNA STANDARDS FOR OTHER APPLICATIONS
DNA standards can be used for optimizing a variety of other applications as it is high-quality genomic DNA with known methylation patterns. This makes them ideal substitutes for precious DNA or problematic sample types when titrating experimental conditions. For example, when optimizing bisulfite library preparation of FFPE DNA, the DNA standards can easily be sonicated or fragmented to be representative of the experimental samples.
The methylated and non-methylated DNA standards have been adapted as controls for numerous methylation assays:
- MethyLight assays 5, 6
- Methylation-sensitive high-resolution melt analysis 7-9
- Pyrosequencing 10, 11
- Methylation arrays 12
- Methylated DNA Immunoprecipitation 13
- Bisulfite sequencing library preparation 14
The Human Methylated & Non-methylated DNA Standards are an example of a validated control set for methylation analysis of human samples. They have been widely used for all the assays mentioned above and much more. Zymo Research also provides a range of other DNA standards that can be used for optimization or quality controls for other assays to help guarantee your data is of the highest quality and ready for publication.
Still unsure of what standards work best for your application? Please email Technical Support ( tech@zymoresearch.com) for help!
DNA METHYLATION STANDARDS PRODUCT GUIDE
| Human DNA Standards | |
| Standards | Suggested Applications |
|---|---|
| Human Methylated & Non-Methylated DNA Set |
|
| Universal Methylated DNA Standard |
|
| Human Methylated & Non-Methylated (WGA) DNA Set |
|
| Human Matched DNA Set |
|
| Mouse DNA Standards | |
| Universal Methylated DNA Standard |
|
| Mouse 5-hmC & 5-mC DNA Set |
|
| Escherichia Coli | |
| E. coli Non-Methylated Genomic DNA |
|
| Artificial DNA Standards | |
| 5-Methylcytosine & 5-Hydroxymethylcytosine DNA Standard Set |
|
| Methylated & Non-methylated pUC19 DNA Set |
|
References:
1. Robertson, K. D., DNA methylation and human disease. Nat Rev Genet 2005, 6 (8), 597-610.
2. Roy, D.; Tiirikainen, M., Diagnostic Power of DNA Methylation Classifiers for Early Detection of Cancer. Trends Cancer 2020, 6 (2), 78-81.
3. Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G., MGMT testing--the challenges for biomarker-based glioma treatment. Nat Rev Neurol 2014, 10 (7), 372-85.
4. Hernández, H. G.; Tse, M. Y.; Pang, S. C.; Arboleda, H.; Forero, D. A., Optimizing methodologies for PCR-based DNA methylation analysis. Biotechniques 2013, 55 (4), 181-97.
5. Olkhov-Mitsel, E.; Zdravic, D.; Kron, K.; van der Kwast, T.; Fleshner, N.; Bapat, B., Novel multiplex MethyLight protocol for detection of DNA methylation in patient tissues and bodily fluids. Sci Rep 2014, 4, 4432.
6. Delgado-Cruzata, L.; Vin-Raviv, N.; Tehranifar, P.; Flom, J.; Reynolds, D.; Gonzalez, K.; Santella, R. M.; Terry, M. B., Correlations in global DNA methylation measures in peripheral blood mononuclear cells and granulocytes. Epigenetics 2014, 9 (11), 1504-10.
7. Ribeiro Ferreira, I.; Darleans Dos Santos Cunha, W.; Henrique Ferreira Gomes, L.; Azevedo Cintra, H.; Lopes Cabral Guimarães Fonseca, L.; Ferreira Bastos, E.; Clinton Llerena, J.; Farias Meira de Vasconcelos, Z.; da Cunha Guida, L., A rapid and accurate methylation-sensitive high-resolution melting analysis assay for the diagnosis of Prader Willi and Angelman patients. Mol Genet Genomic Med 2019, 7 (6), e637.
8. Rajić, J.; Inic-Kanada, A.; Stein, E.; Dinić, S.; Schuerer, N.; Uskoković, A.; Ghasemian, E.; Mihailović, M.; Vidaković, M.; Grdović, N.; Barisani-Asenbauer, T., Infection Is Associated with E-Cadherin Promoter Methylation, Downregulation of E-Cadherin Expression, and Increased Expression of Fibronectin and α-SMA-Implications for Epithelial-Mesenchymal Transition. Front Cell Infect Microbiol 2017, 7, 253.
9. Rawluszko, A. A.; Bujnicka, K. E.; Horbacka, K.; Krokowicz, P.; Jagodziński, P. P., Expression and DNA methylation levels of prolyl hydroxylases PHD1, PHD2, PHD3 and asparaginyl hydroxylase FIH in colorectal cancer. BMC Cancer 2013, 13, 526.
10. Sparago, A.; Verma, A.; Patricelli, M. G.; Pignata, L.; Russo, S.; Calzari, L.; De Francesco, N.; Del Prete, R.; Palumbo, O.; Carella, M.; Mackay, D. J. G.; Rezwan, F. I.; Angelini, C.; Cerrato, F.; Cubellis, M. V.; Riccio, A., The phenotypic variations of multi-locus imprinting disturbances associated with maternal-effect variants of NLRP5 range from overt imprinting disorder to apparently healthy phenotype. Clin Epigenetics 2019, 11 (1), 190.
11. Yu, W.; Qin, X.; Jin, Y.; Li, Y.; Santiskulvong, C.; Vu, V.; Zeng, G.; Zhang, Z.; Chow, M.; Rao, J., Tianshengyuan-1 (TSY-1) regulates cellular Telomerase activity by methylation of TERT promoter. Oncotarget 2017, 8 (5), 7977-7988.
12. Van der Auwera, I.; Yu, W.; Suo, L.; Van Neste, L.; van Dam, P.; Van Marck, E. A.; Pauwels, P.; Vermeulen, P. B.; Dirix, L. Y.; Van Laere, S. J., Array-based DNA methylation profiling for breast cancer subtype discrimination. PLoS One 2010, 5 (9), e12616.
13. Rauen, T.; Grammatikos, A. P.; Hedrich, C. M.; Floege, J.; Tenbrock, K.; Ohl, K.; Kyttaris, V. C.; Tsokos, G. C., cAMP-responsive element modulator α (CREMα) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE). J Biol Chem 2012, 287 (51), 42525-32.
14. Zheleznyakova, G. Y.; Nilsson, E. K.; Kiselev, A. V.; Maretina, M. A.; Tishchenko, L. I.; Fredriksson, R.; Baranov, V. S.; Schiöth, H. B., Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS One 2015, 10 (3), e0121964.





